
|
|
SyMAP System Guide | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The following discusses building a SyMAP v5 database. This document
has been updated as of v5.0.7; see Release Notes for changes.
For release v5.0.8, a document on trouble shooting MUMmer alignments failures has been added.
Contents
Overview and PublicationsSyMAP is a system for computing, displaying, and analyzing syntenic alignments between medium-to-high divergent eukaryotic genomes. It does not work well for very similar genomes or bacterial genomes.Its features include the following (for a pictorial introduction, see the Tour):
C. Soderlund, M. Bomhoff, and W. Nelson (2011)
SyMAP: A turnkey synteny system with application to plant genomes.
Nucleic Acids Research 39(10):e68.
C. Soderlund, W. Nelson, A. Shoemaker and A. Paterson (2006)
SyMAP: A System for Discovering and Viewing Syntenic Regions of FPC maps
Genome Research 16:1159-1168.
SyMAP is freely distributed software, however
if you use SyMAP results in published research, you must cite one
or both of these articles along with any external programs you used within SyMAP such as
MUMmer1,2
Steps for finding syntenyFollow the steps below to get started with SyMAP. Note that each SyMAP application window has a separate Help button providing further details on the use of its functions.
If you are working with FPC files, see the section on FPC.
The jar files have been compiled with Java 1.8, which is upward compatible. For performing large alignments (e.g. 1Gb genomes or more) it is essential to have multiple CPUs, a 64-bit computer, and at least 5Gb of RAM for each CPU that you intend to use. Note that you can set the number of CPUs for SyMAP to use. See MUMmer for information on insufficient memory.
For viewing alignments, CPU and memory needs are typically negligible, unless
you are performing queries on more than 4-5 genomes at once.
To run SyMAP, enter the symap_5 directory and run the command:
MUMmer1,2 (sequence alignment), Blat3 (FPC alignment)
and MUSCLE7 (SyMAP Queries) are provided with the package.
The first time you run SyMAP, it will create the database with information written to the terminal, e.g. Creating database 'symapDemo' (jdbc:mysql://localhost/symapDemo?characterEncoding=utf8). It will check your MySQL variables; if there are any "Suggested" changes, see Trouble Shoot MySQL. It will also check that the provided executables (e.g. MUMmer); if it shows any problems, see MUMmer Executables.
Ordering the demo draftThe remainder of the demo relates to draft sequence, i.e. unanchored shotgun sequence.
The ordering algorithm changes the order of the draft contigs in the database, but does not change the sequence files on disk. However, it writes the following files: 1. File of ordered contigs: It writes the order of the contigs along with whether they should be flipped to a file called /data/seq/Demo_seq2/ordering.csv. 2. Fasta files of ordered sequences: It creates sequence files from the ordered contigs that are flipped when appropriate, which are put into a new project with the suffix "_ordered", as shown on the right. The chromosome names correspond to the order-against project (e.g. Demo_seq2), and the third chromosome is 'chr0' which contains all draft sequences that were not placed.
Further demos:
MySQL and parametersIf your machine does not have MySQL or MariaDB, download and install it. For example, MySQL can be downloaded from dev.mysql.com. On a personal MacOS, simply download the '.dmg' file and following the instructions. On a work server, the system administrator may need to install it.The MySQL installation does not need to be on the machine where you will do the computations or view the results, as long as it is on an accessible network. Once the server is ready, fill out the database parameters in the symap.config file in the main SyMAP directory, as described below. Important Note: The default settings of MySQL are poorly suited for large-scale data storage. You will want to adjust the parameters innodb_buffer_pool_size and innodb_flush_log_at_trx_commit as described in Trouble Shoot MySQL.
Example symap.config. db_name = symapDemo db_server = localhost db_adminuser = root db_adminpasswd = <password> db_clientuser = db_clientpasswd =To use an alternative file than symap.config, use the "-c" command line argument, e.g. >./symap -c symapTmp.configThis is useful if you have multiple SyMAP databases. Runtime and MemoryIf SyMAP runs out of memory, see Trouble Shoot. If MUMmer runs out of memory, see MUMmer.The largest component of SyMAP execution time is in running MUMmer1,2. The time and memory for MUMmer all depends on the size of the genomes. For example, to align rice (12 chromosomes, 370Mb) to maize (10 chromosomes, 2Gb) required 1 hour and 3 minutes using 8 CPUs with 2.3Ghz speed. SyMAP used one CPU per maize chromosome to align the enter rice chromosome against each of the 10 maize chromosomes (i.e. used 10 CPUs). The memory usage of MUMmer is typically 5G per CPU, however it can be as high as 10G for very long or repetitive chromosomes. If MUMmer fails, it is often due to insufficient memory, see the MUMmer document, which explains how to determine the problem and ways around it.
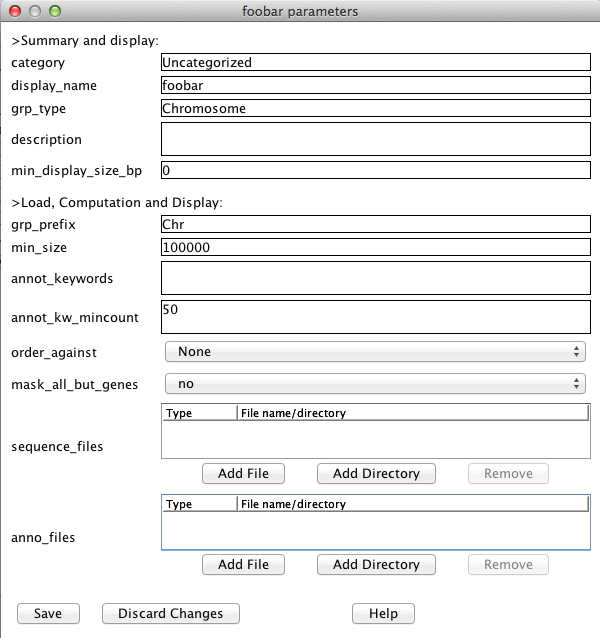
Parameters
After setting the parameters, the project is ready to be loaded.
Preparing the SequencesThe first decision with whole-genome sequence is whether to used repeat masked sequences.
Another masking option which is available if you have gene annotation is to mask out everything but the annotated genes. You can enable the "mask_all_but_genes" option on the Project's Parameters window (shown above); turn it on before doing the alignments. Note that sequence files should be in FASTA format and the name of a sequence is the string immediately following the ">", e.g. >Chr3 Oryza Sativa GAATTCGAATTTGGGTAATGCTAATCAATACAGGTCAAAATCTATGTATTGAGTGGAATATACTGCAAAGTAATTACCTT CTTCCAAAGGAAAGCATTCCTTCTCTCTTGTGGGACTAGCAGATGATCTCGCAGCCAAGACGTGACCACCCAAGGCTCAC ...In this example, the sequence name is "Chr3" and the Chr3 sequence follows. The additional information "Oryza Sativa" is ignored. Three things are important in naming sequences for SyMAP:
Annotation filesAnnotation files should be in gff3 format. The first column (seqid) must exactly match the sequence names in the fasta files. The third column (type) determines how SyMAP uses the entry. Types "gene", "exon", "CDS", "centromere", and "gap" are recognized (other entries are ignored). "CDS" and "exon" are treated equivalently in SyMAP.The last column (attributes) contains "tag=value" pairs describing the annotation. You can set which attributes to use, or use all those occurring more than a certain number of times; open the Project's Parameters window (shown above), look for parameter "annot_keywords". Important: Only the annotation on "gene" entries is shown in the displays or used for searching. For entries "exon", "CDS", "gap", and "centromere", only the coordinates from the gff file are used; the annotation text is not read. NCBI files: A Java script (scripts/ConvertNCBI.class) has been provided that converts NCBI genome fasta files and gff annotation files into the format that works best with SyMAP. See the documentation for instructions. Ensembl files: A Java script (scripts/ConvertEnsembl.class)
has been provided that converts Ensembl genome fasta files and gff3 annotation files
into the format that works best with SyMAP.
See the documentation for instructions.
If you are not ordering the draft sequence,
and if the draft sequence is in too many sequence pieces,
then (1) it takes a long time for the MUMmer comparisons, (2)
the display is very cluttered, and
(3) the blocks display does not work right. Limit the number of sequence pieces
by setting min_size in the parameters window to only load the largest 150 sequences;
there is a script called scripts/lenFasta.pl which
will print out all the lengths; set the min_size to the 150th length. However, even 150 are a lot of blocks to view
so you might want to start with the largest 50, merge them, then repeat.
By default, SyMAP uses the MUMmer 'nucmer' program for self-alignments. Each chromosomes is compared to every other chromosome including itself. The Alignment & Synteny "Parameters" popup has an option to set "Self Args", which only uses the "Self Args" parameter(s) when comparing the chromosome sequence file to itself. In v4.2, the "--maxmatch" parameter was always used, but now it is up to the user as to whether they want to add it to "Self Args" (it can greatly increase the execution time). Reasoning for using "--maxmatch": MUMmer ordinarily seeds its alignments with unique matches, which eliminates the possibility of off-diagonal seeds in the alignment of a chromosome to itself. To overcome this problem, individual chromosome self-alignments can use the MUMmer parameter -maxmatch, which removes the uniqueness requirement at the cost of greatly increased noise. The extra noise is then filtered to a large extent by the default SyMAP filters, but the diagonal squares of the dot plot will still have more noise visible than the off-diagonal.
/data/seq/<project-name>/sequence /data/seq/<project-name>/annotationYou may do one of the following:
The file results of the alignment and synteny computations are is as follows: /data/seq_results/<project-name1>-to-<project-name2>/align /data/seq_results/<project-name1>-to-<project-name2>/finalThe log files are in the /logs directory, see Running MUMmer for details. Alignments and external programsAlignment executablesThe alignment programs are provided in the symap/ext directory. There are executables for 64-bit Linux and 64-bit MacOS. SyMAP will select the correct directory for the machine you are running from, i.e. you do not need to do anything. Typically, these will work just fine; but if they do not, see MUMmer.MUMmer with SyMAP detailsAs of SyMAP v5.0.8, all MUMmer details have been put in a separate document, see MUMmer. This includes trouble shooting when MUMmer fails, and running MUMmer outside of SyMAP.
Working with FPC FilesSyMAP can also align genomes represented by an FPC physical map, by first aligning the BACs using BAC-end sequences or marker sequences. If you plan to work with FPC alignments, the first step is to run the provided demo "Demo-FPC". Align it to "Demo-Seq2" using the same steps as described above, and explore the various displays.Creating an FPC project is the same as for a sequence project except that you choose the type "fpc", and then the Project Parameters window has some different parameters. The Parameters window is where you will enter the FPC file, and your fasta files of marker and BAC-end sequences. Note that the BAC-end sequence names must be exactly the clone names used in FPC, with extension "r" or "f" labeling the strand. In other words, if the FPC map has a clone "a0435B26" then the BES for that clone can be named "a0435B26f" or "a0435B26r". The BES and marker alignments in an FPC project are performed using BLAT3. The running time is typically several times longer than that of MUMmer (described here), but the memory usage is much lower. The default locations are: /data/fpc/<project-name>/<name>.fpc; /data/fpc/<project-name>/bes /data/fpc/<project-name>/mrkThe options for sequence files under directory structure applies to FPC files using these default locations.
Alignment:
The sequences are written to disk*, with gene-masking if desired. In the alignment, one species is "query" and the other is "target". If one project is FPC, that is the query; if both are sequence, the query is the one with alphabetically the first name. The query sequences are written into one large file, while smaller target sequences are grouped into larger fasta files of size up to 60Mb, for more efficient processing in MUMmer. There is an option "Concat" that if unchecked, both query sequences are treated the same as the target; this is useful if the query and target are very large genomes.
Anchor Clustering:
Anchor Filtering:
Synteny Block Detection: * Note that the sequences are re-written from the database to the disk for three reasons: (A) To allow re-grouping for efficiency; (B) To ensure elimination of invalid characters; (C) To mask non-gene regions, if desired. This also ensures that sequences names will match those in the database, and prevents problems caused by moving the source sequences on disk.
2 Marcais, G., A.L. Delcher, A.M. Phillippy, R. Coston, S.L. Salzberg, A. Zimin (2018) MUMmer4: A fast and versatile genome alignment system, PLoS computational biology, 14(1): e1005944. 3 Kent, J. (2002) BLAT--the BLAST-like alignment tool, Genome Research 12:656-64. 4 Krzywinski, M., J. Schein, I. Birol, J. Connors, R. Gascoyne, D. Horsman, S. Jones, M. Marra (2009) Circos: An information aesthetic for comparative genomics. Genome Research doi:10.1101/gr.092759.109. 5 Soderlund, C., Nelson, W., Shoemaker, A., and Paterson, A.(2006) SyMAP: A system for discovering and viewing syntenic regions of FPC maps. Genome Res. 16:1159-1168. 6 Soderlund, C., Bomhoff, M., and Nelson, W. (2011) SyMAP: A turnkey synteny system with application to multiple large duplicated plant sequenced genomes. Nucleic Acids Res V39, issue 10, e68. 7 Edgar, R (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 113. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||


















| Email Comments To: symap@agcol.arizona.edu |