

|
|
|
|
Input files
| Sequence | Annotation | Location of files | Go to top |
The input is one or more FASTA files of sequences (genome, scaffold, contig), with optional GFF formatted annotation file(s).
- As of v5.5.7, most NCBI and Ensembl files can be loaded directly.
See Tested genomes - Load Originals for what NCBI/Ensembl files
did not work and why. Regardless, it is MUCH better to convert using xToSymap.
- xToSymap converts the NCBI and Ensembl files to a format friendly to symap;
i.e. it is superior than directly loading the files;
see NCBI and Ensembl.
If you are using files from another source, see Other.
- The documentation and SyMAP interface assumes a knowledge of the Linux directory (folder) structure.
Sequence files
The sequence file(s) must be FASTA format with one or more sequences. A header line, which starts with a ">", occurs before the sequence, e.g.>Chr01 NC_003070.9 chromosome ccctaaaccctaaaccctaaaccctaaacctctGAATCCTTAATCCCTAAATCCCTAAATCTTTAAATCCTACATCCATG AATCCCTAAATACCTAAttccctaaacccgaaaccggTTTCTCTGGTTGAAAATCATTGTGtatataatgataattttat...The first word (Chr01) is used as the sequence identifier (seqid) in symap. If the seqid is >20 characters, it will be truncated to 20 characters. The second word is the secondary name (NC_003070.9), see Secondary name.
Identifier (seqid)
Important points in naming sequences for SyMAP:| A. | Sequence identifiers (seqid) can only contain letters, numbers, underscores "_", dash "-" or period ".". This is in contrast with names allowed in gff3 files, which allows the seqid to contain the additional characters of [:^*$@!+?|]. The seqid must be renamed if it contains any of these characters. |
| B. | The seqid must exactly match those used in the annotation files (first column), or the annotations will not be loaded. |
| C. | Use a consistent seqid prefix such as "Chr" for all sequences, then set Group prefix to the prefix in project's Parameter panel. |
| D. | If there is not a consistent seqid prefix (e.g. there are chromosomes and scaffolds, each of which has a different prefix), you may leave the Group prefix blank. Make the names short so they will not clutter the display. You may need to rename your sequence, in which case, it must be done in the FASTA and GFF files. |
| E. | The xToSymap NCBI and Ensembl conversion programs will create a short seqids and provide the renaming in both the FASTA and GFF files. One of these programs may also work for files created elsewhere; see Other inputs. |
Masking
Masked sequence:- SyMAP does not perform the repeat-masking so it must be done with another program.
However, you may obtain masked sequences from NCBI or Ensembl.
- NCBI provides soft-masked sequences where the xToSymap NCBI interface provides the option to change it to hard-masked.
- Ensembl provides both soft and hard-masked sequences.
- Masking reduces alignment time and false-positive hits,
but also runs a risk of concealing true hits due to inaccurate masking.
Masking is not really necessary unless the genome is highly repetitive
and those repeats are shared with other genomes being aligned.
- Occasionally, MUMmer fails aligning sequences, which can be resolved by masking the sequence; the SyMAP webpage MUMmer explains MUMmer failures and how to fix them.
Mask genes:
- Another masking option, which is available if you have gene
annotation, is to mask out everything but the annotated genes. You
can enable the Mask non-genes option on the
Pair Parameter window;
turn it on before doing the alignments.
- This is useful if you only care about the gene synteny, you are confident in your GFF file,
and you want very fast MUMmer computations.
- This is also useful for really large genomes, e.g. wheat is 14Gb and could only be processed on a Mac laptop by masking it.
Annotation files
Annotation files should be in gff3 format, which is a tab-delimited file of 9 columns.| Seqid | The first column must exactly match the sequence identifier in the FASTA files. | ||||||
| Type | The third column determines how SyMAP uses the entry.
Only types gene, mRNA, exon, CDS, gap and centromere are recognized. | ||||||
| Attribute | The last column contains "keyword=value" pairs describing the annotation.
| ||||||
| Order | Each mRNA and its exons/CDS must come after the parent gene and before the next gene.
The mRNA must be before its exons/CDS, but it is okay if all the mRNAs for a gene are listed first. Exons are entered in the order they are found; they must be either ascending or descending. |
For example, the following is from a Ensembl gff3 file:
1 araport11 gene 3631 5899 . + . ID=gene:AT1G01010;Name=NAC001;biotype=protein_coding;description=NAC domain containing protein 1 [Source.... 1 araport11 mRNA 3631 5899 . + . ID=transcript:AT1G01010.1;Parent=gene:AT1G01010;Name=NAC001-201;biotype=protein_coding;tag=Ensemb... 1 araport11 five_prime_UTR 3631 3759 . + . Parent=transcript:AT1G01010.1 1 araport11 exon 3631 3913 . + . Parent=transcript:AT1G01010.1;Name=AT1G01010.1.exon1;constitutive=1;ensembl_end_phase=1;ensembl_p... 1 araport11 CDS 3760 3913 . + 0 ID=CDS:AT1G01010.1;Parent=transcript:AT1G01010.1;protein_id=AT1G01010.1 1 araport11 exon 3996 4276 . + . Parent=transcript:AT1G01010.1;Name=AT1G01010.1.exon2;constitutive=1;ensembl_end_phase=0;ensembl_p... 1 araport11 CDS 3996 4276 . + 2 ID=CDS:AT1G01010.1;Parent=transcript:AT1G01010.1;protein_id=AT1G01010.1 etcFor the above example, the xToSymap converted file will contain:
Chr01 araport11 gene 3631 5899 . + . ID=AT1G01010;Name=NAC001;desc=NAC domain containing protein 1;rnaID=AT1G01010.1 (1); Chr01 araport11 mRNA 3631 5899 . + . ID=AT1G01010.1;Parent=AT1G01010;tag=Ensembl_canonical Chr01 araport11 exon 3631 3913 . + . Parent=AT1G01010.1 Chr01 araport11 exon 3996 4276 . + . Parent=AT1G01010.1 Chr01 araport11 CDS 3760 3913 . + 0 Parent=AT1G01010.1 Chr01 araport11 CDS 3996 4276 . + 2 Parent=AT1G01010.1 etcThe following will be loaded into the SyMAP database, where only the exons from the longest mRNA are loaded:
1 gene 3631 5899 + ID=gene:AT1G01010;Name=NAC001;desc=NAC domain containing protein 1 1 exon 3631 3913 Parent=AT1G01010 1 exon 3996 4276 Parent=AT1G01010 etcNote: as of v5.8.6, the xToSymap conversion programs include CDS coordinates in the converted .gff/.gff3 files. To load the CDSs instead of the default exons, change it on the Project Parameter panel.
Location of files
Project name and directory:
- The project-name should be a short descriptive name
containing only alphanumeric characters, periods, underscores and hyphens; do not use symbols like %, $, spaces, etc.
The names are case-sensitive, e.g. you cannot use "Mus" and "mus" for two different projects.
- The project-name is the same as the directory name for the project, i.e.
data/seq/<project-name> - The project-name is used in the data/seq_results directory name and MUMmer output file names.
For example, the alignments between project-names demo_seq and demo_seq2 are in the directory
data/seq_results/demo_seq_to_demo_seq2
and the MUMmer results file are named using it,
demo_seq_f2.demo_seq_f1.mum. - You should not rename this directory after the data is loaded into SyMAP! However, you can define a Display Name in the project's Parameter to be used in the symap displays, which can be change at any time.
Location:
Each project has a directory as follows:
/data/seq/<project-name>
The default location for sequence and annotation files is:
/data/seq/<project-name>/sequence /data/seq/<project-name>/annotationCreate project: Use one of the following ways to indicate to symap where the project's input files are:
- Default: Create these sub-directories under /data/seq and put your files there,
e.g using project-name=foobar:cd data/seq mkdir foobar cd foobar mkdir sequence mkdir annotation
Move your FASTA file(s) to data/seq/<project-name>/sequence (e.g. data/seq/foobar/sequence) and your optional GFF files(s) to data/seq/<project-name>/annotation (e.g. data/seq/foobar/annotation) - Use xToSymap: If you use this program to convert your NCBI or Ensembl files,
it automatically puts the files in the default locations;
see xToSymap.
- Link: Create these sub-directories under /data/seq and use soft links to point to the file locations,
e.g using project-name=foobar:cd data/seq mkdir foobar cd foobar ln -s <location of directory of sequence files> sequence ln -s <location of directory of annotation files> annotation
- Add and define:
- Use the Add project button on the symap interface (lower-left corner; see Manager). This will add the project name to the data/seq directory.
- Use the project's parameter panel to enter the location of the sequences and optional annotation files into the Sequence files and Anno files parameters.
Load Project
For options 1-3, it is not necessary to enter the locations of the files in the project parameter panel since they use the default locations.
|
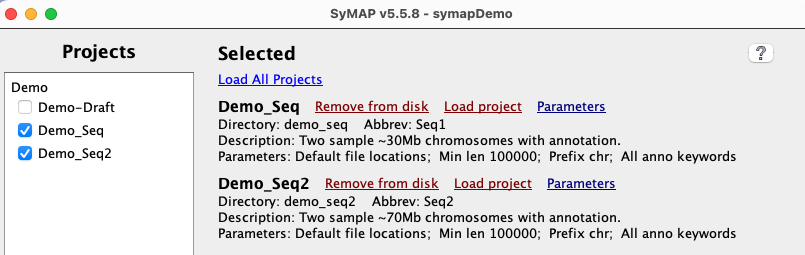
All sub-directories in data/seq are shown on the left-panel under Projects
using its Display name, which is set in the project's parameters.
Select a Project to show it on the right-hand side; the project-name is shown beside the label Directory. |
|
| Load project accesses the files and loads them into the database. Once loaded, the view option will be present. Always check the results before continuing!! | 
|
{kind=link}
The symap program requires the directories to be present (and will create them if necessary), viewSymap does not access the directories.
xToSymap Interface
| Interface | Summarize | Convert | Lengths | Split | Go to top |
xToSymap is in the SyMAP tar file. xToSymap
was last updated v.5.8.6.
|
Project Directory: All options and function buttons will be disabled until a project directory is selected. The option Converted and functions Lengths and Split are disabled when there is no /sequence sub-directory, which is created when Convert is executed. | 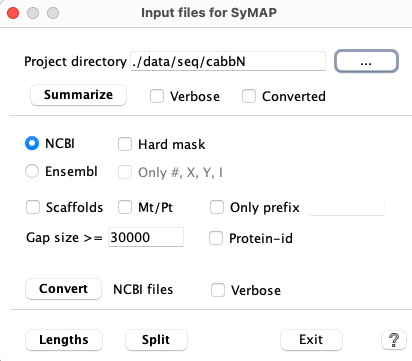
|
| 1. | Summarize | This will output basic statistics. Run this first to make sure your input is valid. |
| 2. | Convert | The NCBI and Ensembl options create the /sequence and /annotation sub-directories with files ready for input to symap. If these sub-directories exist, any existing FASTA and GFF files will first be removed. |
| 3. | Summarize
with Converted | Run this to make sure that the conversion worked as you expected.
This shows the sequence prefixes, and can be used to set the Group Prefix in the symap Project Parameters. |
| Optional. The following run on the files in the /sequence and /annotation sub-directories. | ||
| 4. | Lengths | Run it to see if you need to set a Minimal lengths value in symap Project Parameters. |
| 5. | Split | Splits the converted file genomic.fna and anno.gff into a file per chromosome. |
NOTE: The NCBI and Ensembl files have variations that I may not have accounted for. If Convert does not work as you wish, the files scripts/ConvertNCBI.java and scripts/ConvertEnsembl.java are available for Edit.
Summarize
Summarize will look in the following directories for the FASTA and optional GFF files to process, in the following order (this assumes a project name is cabb):- Directly under the project's directory, e.g.
symap_5/data/seq/CabbE Brassica_oleracea.BOL.dna_sm.toplevel.fa.gz Brassica_oleracea.BOL.59.gff3.gz - The project's ncbi_datasets/data/<sub-directory>, e.g.
symap_5/data/seq/CabbN/data/seq/ncbi_dataset/data/GCF_000695525.1 GCF_000695525.1_BOL_genomic.fna genomic.gff - The project's sub-directories /sequence and /annotation, e.g
symap_5/data/seq/Cabb sequence/ genomic.fna annotation/ anno.gff
The GFF file must end in .gff or .gff3 (with optional .gz).
Summarize with Converted assumes there are converted files.
The following is an example terminal and log file output with Verbose:
------ Summary for ./data/seq/cabbN on 09-Jul-26 ------
Summary file: ./data/seq/cabbN/xSummary.log
Sequence directory: ./data/seq/cabbN
GCF_000695525.1_BOL_genomic.fna File date 23-Jul-2024 14:06
Annotation directory: ./data/seq/cabbN
genomic.gff File date 23-Jul-2024 14:06
### FASTA sequence file(s)
Example header lines
Chr: >NC_027748.1 Brassica oleracea var. oleracea cultivar TO1000 chromosome C1, BOL, whole genome shotgun sequence
Scaf: >NW_013617415.1 Brassica oleracea var. oleracea cultivar TO1000 unplaced genomic scaffold, BOL UnpScaffold00285, whole genome shotgun sequence
Mt: >NC_016118.1 Brassica oleracea mitochondrion, complete genome
Count prefixes
32,876 NW_
10 NC_
Lengths of 3 sequences
Length SeqId
43,764,888 NC_027748.1
52,886,895 NC_027749.1
64,984,695 NC_027750.1
#Seqs<length && >=previous
0<10k 552<100k 41<1M 0<10M 5<50M 4<100M
Bases count for all sequence
A 137,627,834 a 4,845,085
T 137,625,945 t 4,834,468
C 78,312,962 c 2,192,767
G 78,317,437 g 2,192,880
N 43,004,782 n 0
Summary
9 Chromosomes 446,885,882
32,876 Scaffolds 41,708,007
1 Mt/Pt 360,271
32,886 Total 488,954,160
### GFF Annotation file(s)
Gene attribute keywords
44,386 Dbxref 44,386 ID
44,386 Name 569 end_range
7 exception 44,386 gbkey
44,386 gene 44,386 gene_biotype
81 locus_tag 7 part
985 partial 499 start_range
44,305 mRNA's with attribute keyword 'product'
Summary
44,386 Gene from 49,563 (protein_coding)
44,382 mRNA from 56,687 (1st mRNA)
227,191 Exon from 398,922
217,004 CDS from 305,897
Input type: NCBI NC_ chromosome prefix
NCBI 'gene_biotype' keyword; NCBI mRNA 'product' keyword
------ Finish summary for ./data/seq/cabbN/ ------
The last line verifies that it has the NCBI chromosome prefix and attributes used by SyMAP.
After conversion, the summary will be like this log file.
Summarized Verbose
The Ensembl cabbage files Verbose summary log file look like this
Ensembl summary log.
The verbose mode takes longer to execute, but has the following extra information:
- From FASTA:
- Base count has maximum 2.14G bases (instead of 500M).
- Lengths of all chromosomes plus a few of scaffolds, Mt, Pt (instead of the 1st 5 sequences).
- From GFF:
- Some of the header lines from the first GFF file (relevant for Ensembl).
- The first gene, mRNA and exons lines.
- All types from column 3, and all gene attribute biotype values.
Summarize shows a few of the header lines, and with Verbose checked, more are shown. This should aid in determining how to set the options. If this is not enough, execute the following on your FASTA file:
zgrep ">" [use your FASTA file name] | more
Convert
|
1. Select NCBI or Ensembl.
2. Select the options you want as described below. 3. Select Convert NCBI/Ensembl NCBI and Ensembl both have their own general rules for identifying chromosomes, scaffold, etc from the FASTA header line for each sequence; see NCBI and Ensembl. Chromosome sequences will use 'Chr' for the prefix of the seqid unless Scaffolds is selected; additional prefix rules are provided below. | 
|
The following options are mainly for FASTA files that do not follow the NCBI/Ensembl general rules for header lines (see Exceptions):
| Option | Description | Default if not selected |
| Hard mask | (NCBI only) NCBI genome sequences are typically soft-masked, where this option changes it to hard masked | Leave as soft-mask |
| Only #,X,Y,I | (Ensembl only) Most Ensembl FASTA header lines specify the chromosome number, X, Y or Roman numeral. Only these sequences will be written. | Any that have 'chromosome' on their header line. |
| All Seqs1 | Load all sequences. | Ignore un-identified sequences |
| Scaffolds2 | Any sequence identified as scaffold will be written to the FASTA file. Chromosomes will be given the prefix 'C' and scaffolds will be given the prefix 's'. | Ignore scaffolds |
| Mt/Pt | Mt/Pt chromosomes will be included in FASTA and GFF. Only the first occurrence will be included. The prefix of 'Mt' or 'Pt' will be used. | Ignore Mt/Pt |
| Only prefix3 | Only sequences with the specified prefix will be processed. | None |
| Gap | A annotation file called gap.gff is created, and any sequence of n's greater than this number is considered a gap and the coordinates are written to this file. | 30000 |
| Verbose | Print extra information, e.g. see verbose log | No extra information |
1All Seqs selected: If Scaffolds and/or Mt/Pt are also selected, their prefix rules will be used. Otherwise, any sequence not identified as 'chromosome' will be prefixed with 'Seq'.
2Scaffold selected: If there are 100's of scaffolds, limit the input on the symap Load project by setting Minimal length in the project's Parameters.
3If Only prefix is not blank, sequences are first filtered out if the seqid does not start with the prefix. Then, the rest of the Rules are applied as described by NCBI and Ensembl.
It is HIGHLY recommended that you run Summarize on the Converted sequences to determine if the output is what you expected.
Lengths
The FASTA files must be in the project's /sequence sub-directory (e.g. symap_5/data/seq/cabbN/sequence) for this to work. It reads the FASTA file(s), prints out the names and lengths of all sequences, then writes a summary.If your file has many small scaffolds and you just want to process the large ones, the Length output will help you decide what value to use as the Parameter Minimum length values. For example, the following is output from Length:
Values for parameter 'Minimal length' (assuming no duplicate lengths): #Seqs Minimum length 10 550,871 20 193,719 30 152,041 40 122,531 50 98,180 60 85,914 70 70,524 80 65,697 90 61,049 100 58,280If you set the Minimal length at 550,871, SyMAP will process the 10 largest sequences.
Split
The input files are:sequence/genomic.fna annotation/anno.gffThe function will do the following:
- Split genomic.fna into a separate file per chromosome, and put all other sequences in scaf.fna. The genomic.fna file is deleted when done.
- Split anno.gff into a separate file per chromosome, and put all other sequences in scaf.gff. The anno.gff file is deleted when done.
General
| Tested genomes | Load original files | Secondary name | Editing the convert scripts | Go to top |
Tested genomes
In June 2026, the scripts were tested on the following species (most were downloaded in 2024), which shows the variation in NCBI/Ensembl files that I have come across:| Genome | NCBI | Ensembl | Sequence bp in file | Note |
|---|---|---|---|---|
| Arabidopsis thaliana (thale crest) | Chr 1-5 | Chr 1-5 | 119M | |
| Brassica oleracea (wild cabbage) | C1-C9 | C1-C9 | 489M | C prefixes chr# |
| Oryza sativa (rice) | Chr 1-12 | Chr 1-12 | 386M | |
| Prunus persica (peach) | G1-G8 | G1-G8 | 228M | G prefixes chr# |
| Prunus yedoensis | Scaffolds 4015 | N/A | 319M | Draft |
| Caenorhabditis elegans (worm) | Chr I,II,III,IV,X | Chr I,II,III,IV,X | 100M | |
| Bemisia tabaci (whitefly) | Scafs 19,750 | Contigs 227 | 615M | |
| Danio rerio (zebrafish) | Chr 1-25 | Chr 1-25 | 52,633M | Contains 930 ALT CHR |
| Oryctolagus cuniculus (rabbit) | Chr 1-21, X | Chr 1-21, X | 2,841M | |
| Homo Sapiens (human) | Chr 1-22, X, Y | Chr 1-22, X, Y | 3,298M |
Most FASTA files only use a number after the word 'chromosome'. However, some FASTA files prefix the number with a letter.
For example, Cabbage had 'C' before its chromosome number, e.g.
>NC_027748.1 Brassica oleracea var. oleracea cultivar TO1000 chromosome C1, BOL...
so the chromosome names will be C1, C2, etc.
See Tested datasets and timings for the Alignment&Synteny compute times for a range of species pairs.
xToSymap - exceptions to the defaults
The above downloaded genomes were all converted with the xToSymap defaults, with the following exceptions:
- Ensembl Danio rerio (zebrafish): The 100's of ALT sequences appear as valid chromosomes as
the headers contain the word 'chromosome', e.g.
>1 dna_sm:chromosome chromosome:GRCz11:1:1:59578282:1 REF
>CHR_ALT_CTG8_1_12 dna_sm:chromosome chromosome:GRCz11:CHR_ALT....
In order not to include these, the Only #,X,Y,I option was used. - Ensembl Bemisia tabaci (whitefly): The words 'chromosome' or 'scaffold'
were NOT part of the header lines. The header line ID prefix was 'Contig', e.g.
>Contig14 dna_sm:primary_assembly primary_assembly:ASIAII5_n227_616Mb:Contig14:1:53342825:1
The summary showed that all sequences had the same prefix, so All Seqs could be used. Alternatively, the Prefix only=Contig could be set. Either way, the output will use the sequence ID prefix 'Seq'.
Since there were 227 contigs, Lengths was used to determine a Minimal length to enter on symap project Parameters. - NCBI Bemisia tabaci (whitefly): The file only had scaffolds, so including Scaffolds was necessary.
Since there were 19,750 scaffolds, the Lengths was used to determine a Minimal length
to enter on symap project Parameters.
- NCBI Prunus yedoensis:
The words 'chromosome' or 'scaffold'
were NOT part of the header lines. The header line ID prefix was 'PJQY' (not the NCBI prefix of 'NC_'), e.g.
>PJQY01000001.1 Prunus yedoensis var. nudiflora Py_C0000, whole genome shotgun sequence
The summary showed that all sequences had the same prefix, so All Seqs could be used. Alternatively, the Prefix only=PJQY could be set. Either way, the output will use the sequence ID prefix 'Seq'.
Input variations: There are probably more variations then shown here, which may possibly be handled with the available options. If not, you may want to edit the script to tailor it (see Edit). Alternatively, you can contact me (cas1@arizona.edu) and I will make the edit for you (it allows me to add the your variation to xToSymap).
Load original files
To load the originals files into SyMAP, the following was done:
- Create /sequence and move the FASTA file into it.
Create /annotation and move the GFF file into it. - Run the xToSymap Summarize and Lengths functions.
- For Ensembl, a Minimal length may need to be set to only load the sequence >= the smallest chromosome length.
- For NCBI, set the Group prefix to 'NC_' to only load the chromosomes.
For example, for species such as Homo Sapiens, if Group prefix or Minimal length were NOT specified, over 500 sequences were loaded. With Ensembl, it also had a problem with duplicate entries because it has scaffold names such as HSCHR19KIR_CA01-TB04_CTG3_1 and HSCHR19KIR_CA01-TB01_CTG3_1, where the name was truncated at the "-", hence, HSCHR19KIR_CA01 is a duplicate name.
- Ensembl Danio rerio (zebrafish): It could not be loaded without conversion. It has 1922 sequences where most of them had prefix "ALT", the word 'chromosome' in their header line, and were the size of their REF chromosome, so setting a Minimal length did not exclude them. And there was no unique chromosome ID prefix to use as the Group prefix in symap.
Some problems with the original files: (there are probably more)
- Genes are not restricted to protein-coding. The 1st mRNA will be loaded instead of the longest.
- Numerous Queries functions fail if there is a '.' in the original seqid.
- There may be excessive attributes in the Queries table.
- 2D and other displays will likely have chromosomes names written on top of each other due to their length.
Secondary name
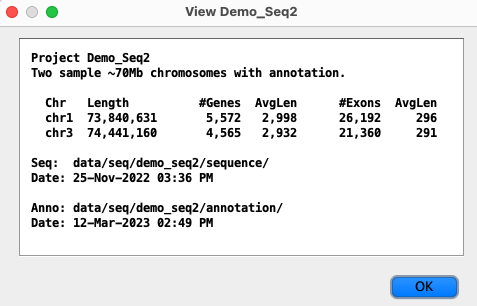
A secondary name can be assigned during the symap Load Project action. If a sequence header has between 2-3 words, the 2nd word will be used as the secondary name as long as it is <=20 characters. The following example shows the header line of an NCBI xToSymap converted file.>Chr01 NC_003070.9 chromosome
| The symap Load Project will make
Chr01 the unique identifier and NC_003070.9 will be its secondary name.
This secondary name will be shown on the View popup.
Ensembl xToSymap converted files do not have a secondary name. | 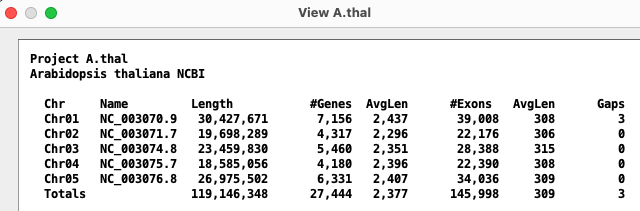
|
Original NCBI File: An example header line will be:
>NC_003070.9 Arabidopsis thaliana chromosome 1 sequenceIn this case, NC_003070.9 will be its identifier and "Chr01" will be its secondary name. The secondary name is only assigned if the value "chromosome " or "scaffold " exists on the header line.
Original Ensembl File: Generally no secondary name is assigned. However, in some cases it does assign one incorrectly, which will look weird in View.
Editing the convert scripts
- The convert scripts are in the symap_5/scripts directory called
convertNCBI.java and convertEnsembl.java.
- Move the one you want to change to the symap_5/data/seq directory, as
it must be run from there.
- Once you make your changes, execute: javac ConvertNCBI.java
You will need to have JDK installed to use the javac command. - To run your compiled script: java ConvertNCBI <project directory name>
These scripts are simply written, using only standard operations found in all major languages.
| Go to top |
Email Comments To: cas1@arizona.edu